 English
English
 German
German
 French
French
 Italian
Italian
 Spanish
Spanish
 Portuguese
Portuguese
 Chinese
Chinese
 Lithuanian
Lithuanian
1.1.4.2 Pyoderma gangrenosum
ICD-11
EB-21
Synonyms
Dermatitis ulcerosa; phagedenic ulcer.
Read more
Dermatitis ulcerosa, Phagedenic pyoderma, phagedenic ulcer.
Epidemiology
Incidence: 3 -10 per million per year. Peak of incidence: 20 to 50 years. Women slightly more often affected than men. Men are more commonly affected in malignancy-associated PG.
Read more
General incidence: 3 -10 per million per year. Peak of incidence occurs between the ages of 20 to 50 years. Women slightly more often affected than men. Men are more commonly affected in malignancy- associated PG.
30-75% of patients with PG have a second underlying immune-mediated disease, most commonly inflammatory bowel disease (IBD) (17-20 %), rheumatoid arthritis (12%) and hematological malignancies (4-8 %).
Definition
Pyoderma gangrenosum (PG) is a rare occurring, chronic, and painful neutrophilic dermatosis that presents with rapidly developing, painful skin ulcers presenting with violaceous undermined borders and peripheral erythema.
Read more
Pyoderma gangrenosum (PG) is a rare, recurrent, chronic and painful neutrophilic dermatosis that presents with rapidly developing, painful skin ulcers presenting with violaceous, undermined borders and peripheral erythema.
Aetiology & Pathogenesis
More than 50% of patients with PG have an associated systemic disease, including:
- Inflammatory bowel disease (IBD) (40%). No correlation withn the severity of IBD.
- Arthritis (12%): Seronegative. Rheumatoid arthritis and ankylosing spondylitis.
- Hematologic malignancies (7%). Most commonly acute myelogenous leukemia, but a broad spectrum of lymphoproliferative disorders.
- Drugs: Interferon-α2b, G-CSF.
- Autoinflammatory diseases:
- PAPA syndrome characterized by destructive polyarthritis, severe nodulo-cystic acne and PG. - PASH syndrome (PG, acne and hidradenitis suppurativa)
- PAPASH syndrome (pyogenic arthritis, PG, acne and hidradenitis suppurativa) 6- Overlap between PG and Sweet’s syndrome: Usually observed in patients with myeloproliferative disorders.
Read more
More than 50% of patients with PG have an associated systemic disease, including:
Inflammatory bowel diseases (Crohn’s disease and ulcerative colitis) are the systemic diseases most frequently reported in association with PG. High relative risk (odds ratio) of a patient with IBD to develop PG is 29.2, in contrast, only 0.75% of patients with inflammatory bowel diseases had PG. There is no association between the severity of inflammatory bowel disease and the presence of PG.
Arthritis: Most frequently seronegative arthritis of a single, large joint. Classical forms of rheumatoid arthritis (RA) and ankylosing spondylitis are also associated.
Hematologic malignancies (7%). Most commonly acute myelogenous leukemia, but a broad spectrum of lymphoproliferative disorders including monoclonal gammopathies, leukemia, lymphoma, and myelodysplastic syndromes have been described in association with PG.
Drugs: Interferon-α2b, isotretinoin, G-CSF in patients with hematological disorders, propylthiouracil, tyrosine kinase inhibitors (sunitinib, imatinib and gefitinib), and tumor necrosis factor-α inhibitors (infliximab, adalimumab), rituximab.
Autoinflammatory diseases:
PAPA syndrome is an autosomal dominant inherited disease characterized by non-axial destructive polyarthritis, PG and severe cystic acne. PAPA syndrome is caused by mutations in the PSTPIP1 gene involved in regulation of inflammatory response.
PASH syndrome (PG, acne and hidradenitis suppurativa) and
PAPASH syndrome (pyogenic arthritis, PG, acne and hidradenitis suppurativa) are other syndromic forms of PG.
Overlap between PG and Sweet’s syndrome: Usually observed in patients with myeloproliferative disorders.
The pathogenesis of PG is unknown. Trauma (pathergy) may induce the development or worsen previous lesions PG lesions (e.g. biopsy).
Release of cytokines (IL-36, IL-8) and danger signals that can enhance innate immune responses.
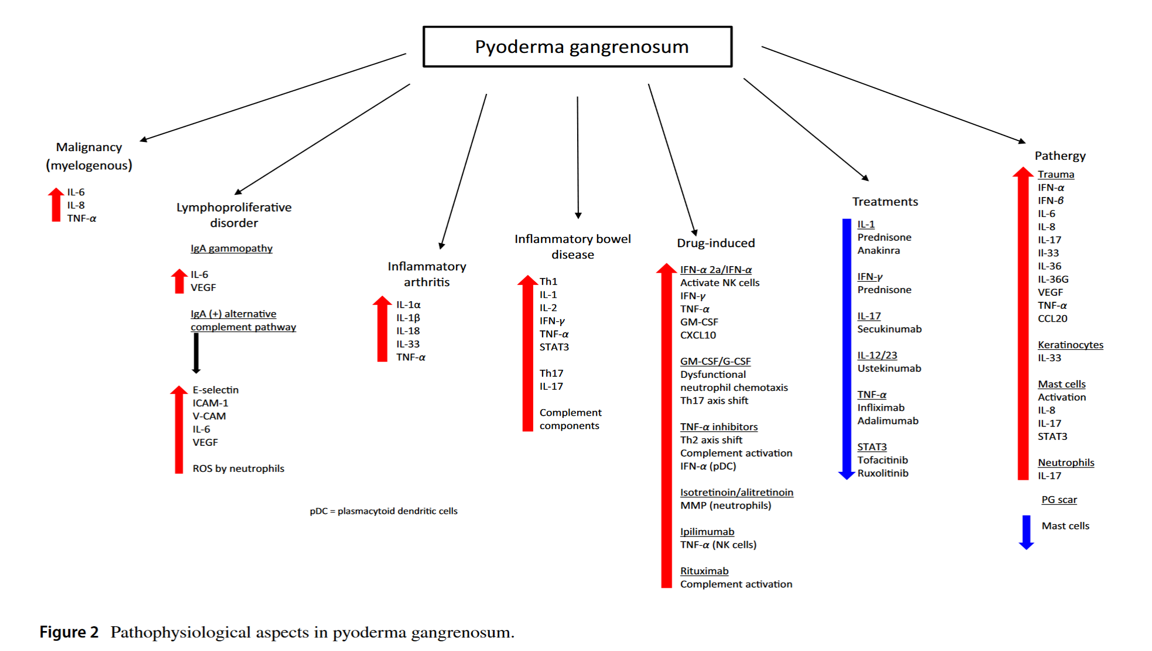
Signs & Symptoms
Presents with a tender nodule, plaque, or sterile pustule that enlarges progressing to sharply marginated painful ulcers with central necrosis, undermined violaceous borders and surrounding erythema and / or bullous lesions. The ulcer usually increases in size and depth (phagedenism).
Solitary lesion or multiple simultaneous recurrent lesions. Tends to develop at areas of previous trauma including surgery (pathergy phenomenon) (25-50%). As the lesions heal, cribiform or wrinkled (cigarette paper-like) scars often develop.
Extracutaneous sterile neutrophilic infiltrates can be observed in the bones, liver, lungs, pancreas, spleen, kidneys, and central nervous system.
Read more
Lesions of PG presents with a tender nodule, plaque, or sterile pustule that enlarges, over a course of days, progressing to sharply demarcated painful ulcers with central necrosis. The lesions have undermined violaceous borders and a surrounding zone of erythema. Central necrotic areas with hemorrhagic or purulent exudates develop. The ulcer usually increases in size and depth or extends through the appearance of new peripherally located pustules.
PG may be a solitary lesion but also may manifest as multiple simultaneously recurrent lesions. In 25- 50% of cases, PG arises at areas of trauma (pathergy). As the lesions heal, cribiform or ‘sieve-like’ atrophic scars or wrinkled (cigarette paper-like) scars develop.
Extracutaneous sterile neutrophilic infiltrates can be observed in the bones, liver, lungs, pancreas, spleen, kidneys, and central nervous system of patients with PG.
Localisation
The pretibial area (shins) are most commonly affected but other parts of the skin and mucous membranes are also affected.
Read more
The lower legs (pretibial area) are most commonly affected but other parts of the skin and mucous membranes including breast, hand, trunk, head and neck and peristomal skin may also be involved.
Classification
Different clinical variants of PG have been defined:
- Ulcerative (classical) PG: Most commonly occurs at sites of trauma. Associated disorders: IBD, hematological malignancies, rheumatoid and seronegative arthritis and monoclonal gammopathy.
- Bullous PG: Painful bulla(e) that can progress to erosion and/or ulcer. Associations: Myeloproliferative disorders (70%) and IBD.
- Pustular PG: Pustules with erythematous borders. Association: IBD.
- Vegetating PG: low-growing, non-purulent, superficial ulcer; borders are not undermined. No associated disorders.
- Peristomal PG: Papules that erode into ulcers adjacent to the stoma. Associations: IBD, enteric malignancy, connective tissue disease and monoclonal gammopathy.
- Postoperative PG: Erythema at the surgical site, wound dehiscence or painful ulcerations that coalesce. Abdominal and breast surgery.
Read more
Different clinical variants of PG have been defined:
Ulcerative (classical) PG: Most commonly occurs at sites of trauma, frequently on the anterior lower extremities. Associated disorders: IBD, hematological malignancies, rheumatoid arthritis, seronegative arthritis and monoclonal gammopathy.
Bullous PG: Painful bulla/e that can progress to erosions and/or ulcers. Increased incidence on the face and the dorsum of the hands. Overlap with Sweet’s syndrome. Subcorneal, subepidermal and intra-epidermal bullae with dermal neutrophilic infiltrate and microabscess formation. Associations: Myeloproliferative disorders (70%) and IBD.
Pustular PG: Pustules with erythematous borders. Association: IBD.
Vegetating (granulomatous superficial) PG: Low-growing, non-purulent, superficial ulcer; borders are not undermined. Head and neck. Histologically: Palisading granulomatous reaction. No associated disorders.
Peristomal PG: Papules that erode into ulcers with undermined borders adjacent to the stoma. Dermal neutrophilic infiltrates with granulation tissue. Associations: Inflammatory bowel disease (IBD), gastrointestinal malignancy, connective tissue disease and monoclonal gammopathy.
Postoperative PG: Erythema at the surgical site, followed by wound dehiscence or painful ulcerations that coalesce. Commonly associated with abdominal and breast surgery.
Laboratory & other workups
Depends on the cause of the disease.
PG diagnostic workup includes a complete blood count with peripheral blood smear, biochemical survey, serum and urine protein electrophoresis, syphilis serologic tests and immunological tests (pANCA, cANCA). Skin biopsy for microbiological studies (bacterial, fungal and mycobacterial) should be obtained.
Additional tests may be indicated in cases with concomitant joint or gastrointestinal symptoms or suspicion of underlying hematologic malignancies. Age-appropriate cancer screening in selected cases.
Read more
Occasionally peripheral leukocytosis with neutrophilia and elevated markers of inflammation (erythrocyte sedimentation rate and C-reactive protein).
Laboratory tests are performed to evaluate for associated disorders rather than to establish the diagnosis of pyoderma gangrenosum.
Dermatopathology
The histopathology of PG is nonspecific and changes with the stage of the lesion. Dermal edema, suppurative inflammation and sterile abscess formation. The initial lesions show a deep suppurative folliculitis like pattern with dense neutrophilic infiltrate.
Read more
The histopathology of PG is nonspecific and changes with the stage of the lesion. Dermal edema, suppurative inflammation and sterile abscess formation. At the periphery of the lesion, perivascular or perifollicular lymphoid infiltrates may also be present. Additional staining of the biopsy sample is required in order to rule out bacterial and fungal infections.
The initial lesions show a deep suppurative folliculitis with dense neutrophilic infiltrate. Focal leukocytoclastic vasculitis changes may be present. PG with necrotizing granulomatous inflammation has been observed.
Course
Variable. Chronic, relapsing or self-limiting disease. From mild to aggressive with intense morbidity.
Complications
Secondary infection, tissue destruction, intractable pain. Impact on quality of life (depression). Complications secondary to long term immunosuppressive therapy.
Diagnosis
Diagnosis of PG is based on history of an underlying disease, typical clinical presentation, histopathology, and exclusion of other diseases that would lead to persistent painful ulcers.
Read more
Diagnosis of PG is based on history of an underlying disease, typical clinical presentation, histopathology, and exclusion of other diseases that would lead to persistent painful ulcers. An Infectious disease should be ruled out. An additional skin biopsy for microbiological studies (bacterial, fungal and mycobacterial cultures, viral or other micro-organism PCR amplification) should be obtained.
PG diagnostic workup includes a complete blood count with peripheral blood smear, biochemical survey, serum and urine protein electrophoresis, stool sampling for occult blood and parasites as well as syphilis serologic tests and immunological studies (rheumatoid factor, ANA, ENA, antiphospholipid antibodies, pANCA, cANCA) for autoimmune diseases.
Additional tests may be indicated in cases with concomitant joint or gastrointestinal symptoms including radiological imaging and/or a colonoscopy to rule out inflammatory arthritis and ulcerative colitis, respectively. Peripheral smear, and bone marrow aspiration or biopsy should be performed, if indicated, to evaluate for hematologic malignancies. Age-appropriate cancer screening in selected cases.
Major criteria
Rapid progression of a painful, necrolytic cutaneous ulcer with an irregular, violaceous, and undermined border
Exclusion of other causes of cutaneous ulceration
Minor criteria
History suggestive of pathergy or clinical findings of cribriform scarring
Systemic diseases associated with PG
Histopathologic findings
Treatment response to systemic steroids
Recently, some new diagnostic criteria for PG have been proposed: PARCELSUS and Delphi diagnostic criteria.
Differential Diagnosis
The differential diagnosis should include a wide range of diseases manifested as chronic and persistent cutaneous ulcers:
Vascular occlusive or venous disease, vasculitis (rheumatoid arthritis, ANCAs-related vasculitis, Behçet's disease etc.), hypercoagulable states (anti-phospholipid syndrome), malignant neoplasms (lymphomas, leukaemia), infectious diseases (ecthyma, deep mycoses, atypical mycobacterial infections, late syphilis, necrotizing cellulitis), exogenous tissue injuries (facticial panniculitis), insect or spider bites, and drug reactions (pustular drug reactions). Pemphigus vegetans.
Read more
The differential diagnosis should include a wide range of diseases manifested as chronic and persistent cutaneous ulcers:
Vascular occlusive or venous disease (including calciphylaxis).
Vasculitis (rheumatoid arthritis, ANCAs-related vasculitis, Behcet’s disease, etc.).
Hypercoagulable states (anti-phospholipid syndrome).
Malignant neoplasms (lymphomas, leukaemia).
Infectious diseases (ecthyma, streptococcal synergistic gangrene, deep mycoses, atypical mycobacterial infections, late syphilis, deep viral herpetic infections, necrotizing cellulitis).
Exogenous tissue injuries (factitial panniculitis), insect or spider bites, and
Drug reactions (pustular drug reactions).
Primary cutaneous lymphomas
Prevention & Therapy
Oral corticosteroids (0.5–1 mg/kg/day) are the mainstay of treatment for rapid control of the disease and, in other cases, ciclosporin may be used as a steroid-sparing agent either alone or in combination with corticosteroids.
In resistant cases, the combination of steroids with different immunosuppressants or steroid-sparing agents such as dapsone, colchicine, azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide, clofazimine, thalidomide, tacrolimus, sulfasalazine and minocycline have been proposed.
Anti-tumor necrosis alpha therapy (infliximab, etanercept, adalumimab, certolizumab), could be prescribed in cases of PG associated with IBD. Other systemic treatments include high doses of intravenous immunoglobulin (IVIG), IL-1 and IL-23 inhibitors (ustekinumab), anti-IL-23 (tildrakizumab) anti-IL-17 (secukinumab) and IL-1 antagonists (anakinra, canakinumab, gevokizumab),and small molecules (JAK-STAT inhibitors).
Appropriate wound dressings of different types according to stage of ulcer. Topical and intralesional treatments (corticosteroids, calcineurin inhibitors) may be used as adjuvant therapies.
Read more
A primary prevention is not possible.
Oral corticosteroids (0.5–1 mg/kg/day) are the mainstay of treatment for rapid control of the disease and, in other cases, cyclosporine A may be used as a steroid-sparing agent either alone or in combination with corticosteroids. In severe and recalcitrant cases, pulsed intravenous methylprednisolone should be considered.
Combinations of steroids with cytotoxic drugs are used in resistant cases. The combination of steroids with dapsone or different immunosuppressants or steroid-sparing agents such as colchicine, azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide, clofazimine, thalidomide, tacrolimus, sulfasalazine and minocycline has been proposed.
Anti-tumor necrosis alpha therapy (infliximab, etanercept, adalumimab, certolizumab pegol), could be prescribed in cases of PG associated with IBD. Other systemic treatments include high doses of intravenous immunoglobulin (IVIG), biologic drugs such as IL-12 and IL-23 inhibitors (ustekinumab), anti-IL-23 (tildrakizumab) anti-IL-17 (secukinumab) and IL-1 antagonists (anakinra, canakinumab, gevokizumab), and small molecules (JAK-STAT inhibitors) or complement Factor C5a inhibitor vilobelimab (IFX-1) have been used with variable success.
Topical treatments including superpotent corticosteroids and calcineurin inhibitors (tacrolimus) and, intralesional therapy (triamcinolone, methotrexate) may be used as adjuvant therapies in conjunction with systemic agents. Topical therapy with wound dressings, skin transplants and the application of bio- engineered skin are useful in selected cases.
Special
PG is included within the spectrum of neutrophilic dermatoses.
Neutrophilic dermatoses are a heterogeneous group of inflammatory skin disorders characterized histologically by a neutrophilic infiltrate with no evidence of underlying infection or vasculitis.
Prognosis
Unpredictable clinical course: from a precipitous onset with rapid spread to a more indolent pattern. Frequent relapses. Potentially life-threatening disease.
Read more
The clinical course of PG is unpredictable—from a precipitous onset with rapid spread to a more indolent pattern. Frequent relapses. Potentially life-threatening disease. In some series with a mortality rate of up to 30%.
Diagnostic algorithm
Review Articles
References
This website uses cookies!
We use cookies to tailor our content to your needs and continuously improve our website. You can decide which cookies you want to allow. Detailed information about the cookies we use can be found in our Privacy Policy and Cookie Settings. You can withdraw your consent at any time.
Comments
Be the first one to leave a comment